Clinical researches are a stepping stone to life-saving medical discoveries. The success of such trials leads to the advancement of medical care. Upon successful clinical research studies, the doctors discover more accessible and less harmful alternatives to existing treatment and develop vaccines or drugs with safer aftereffects.
At certain stages of the research study, researchers require volunteers to be human subjects to test and evaluate the actual effect of the supposed treatment. According to FDA regulations, anyone willing to participate in the exempt research studies must undergo an informed consent process.

Build Your Document
Answer a few simple questions to make your document in minutes
Save and Print
Save progress and finish on any device, download and print anytime
Sign and Use
Your valid, lawyer-approved document is ready
Signing an informed consent document is required in various situations and is widely used in medical settings such as providing treatment, operations, invasive procedures, or conducting a clinical investigation.
Informed consent for exempt research study enrollment requires a researcher to educate the participant about the goals, duration period, possible risks, and benefits of participation. Part of the required consent elements, such as explanation, can be conducted verbally through the conversation with a participant, but the essential details must be duplicated in informed consent documents.
The consent must be given knowingly and voluntarily upon evaluating all possible threats and consequences. Only after the participant fully comprehends the terms and purpose of the study and the personal health risks that may happen during trials can they sign a written consent document.
Informed consent is a multi-stage process that leads to the final stage of signing consent forms. Failing to perform each stage of the informed consent process properly may cause legal ramifications. The researcher must educate the potential participant with adequate information to comprehend the possible consequences of the given or refused consent and make an educated decision.
Four main elements of informed consent are required to conduct the consent process properly: patient’s competency (ability to grant permission), legibility, information about the research, and consent documentation.
The informed consent forms with a purpose for enrollment in research trials must contain the following sections:
In specific cases, a researcher may specify additional elements in the informed consent forms, including unforeseeable risks management or involuntary termination. It is strongly advised to make a written document as specific as possible and list all things that may arise during the research projects.
When the clinical trials’ prospective participants are the patients who have no decision-making capacity, their legal representative can grant permission to participate in alternative treatments or research studies on behalf of the person who is unable to do so. These may be incompetent patients or patients in an unconscious state.
If the research subject is a minor, the volunteer must have parental permission to enroll and the child’s consent or agreement. Unless the parent holds full custody over the child or the other parent is deceased or incapable, permission from both parents is required.
The peculiarity of clinical trial informed consent is that it doesn’t end after signing the consent document. The participant can ask researchers for clarifications regarding the studies during and after the completion of trials.
Besides, participants have a right to withdraw their consent due to health risks or other reasons during the research. However, the participant may also face the ramifications upon premature termination. These ramifications are usually described in the informed consent forms.
On the other side, the researcher may also eliminate the participant before the study’s completion due to any plausible reason, such as a change of eligibility criteria or the participant’s failure to comply with the prescribed procedures.

An informed consent document is a legal form that must include all elements required by federal regulations. You can use the informed consent form template available on our website. Be sure to stick to these steps while completing the form.
Step 1. Provide the date and participant’s name
In the document’s opening section, provide the date when the document is being completed and the participant’s full name who gives consent to participate in the study.

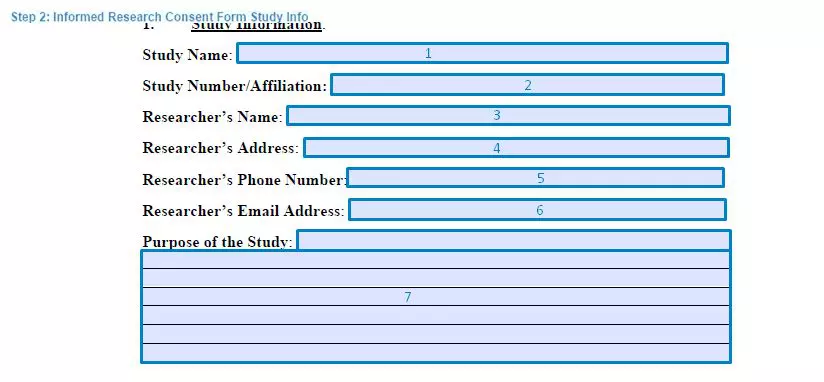
Step 2. Specify the study information
Next, the document must specify the extensive details regarding the research study. It includes the information of the affiliation, institution, and contact details of the researcher that conducts the study. All the fields include:
Step 3. Indicate the participants number
It is also requested that the consent form display the number of participants who will undergo the study procedures.

Step 4. Clarify the study date/hours
Informed consent forms must clarify the period during which the research is conducted. Include the days when the research begins and ends. Also, the document must usually tell the approximate period the participant is expected to spend on the study, calculated in hours, days, or weeks.

Step 5. Specify the participant criteria
In this section, the researcher must describe the criteria to evaluate the eligibility of human subjects.

Step 6. Indicate the purpose of the study
The study participants have the right to be informed regarding the goals and purposes of the study.

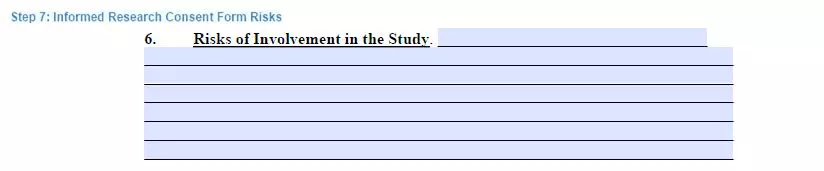
Step 7. Describe the risks
Clarify the potential threats to a person’s health condition. It is not mandatory to describe the risks in full detail. Still, it is necessary to tell the volunteer about any possible irreversible changes to a participant’s health for them to make a proper conscious and educated choice.
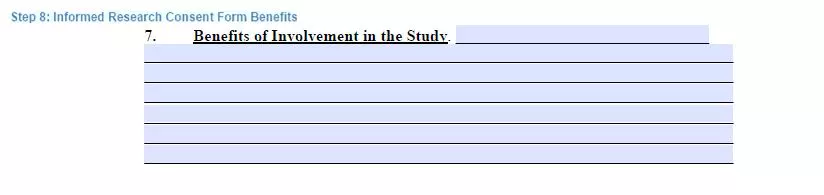
Step 8. Explain the benefits
Stating the benefits of the study can stimulate participants to be obedient and diligent during the research period. That is why stating them in the document is essential.
Step 9. Define the compensation
If the study is funded (by sponsors or governments), it can apply the stimuli in reward payment for participation. The amount of money given to the participants must be documented if applicable.

Step 10. Provide the cost of involvement
For some clinical tests or research studies, the participant must pay money for medication or treatment. If any cost is applied, you need to state it in the document.

Step 11. Explain the confidentiality
The researcher can ask participants to sign a non-disclosure statement to prevent them from leaking any information about the study, its methods, or any other details about the research process.

Step 12. Get the full name and signature
Participants who consent to participate in the trials must provide their full names and sign the written form.
