




Should you desire to fill out medwatch form 3500, you won't have to install any sort of applications - simply use our online tool. In order to make our editor better and simpler to utilize, we continuously implement new features, with our users' feedback in mind. If you are seeking to get going, this is what it's going to take:
Step 1: Click on the "Get Form" button above. It will open up our tool so that you can start filling in your form.
Step 2: As you launch the tool, you'll see the form all set to be completed. Apart from filling in different fields, it's also possible to do many other actions with the Document, including adding custom words, changing the initial textual content, inserting illustrations or photos, signing the document, and much more.
As for the fields of this particular form, here's what you should know:
1. The medwatch form 3500 will require particular details to be inserted. Make certain the following blanks are filled out:

2. Once your current task is complete, take the next step – fill out all of these fields - eqssqauO s, M qOq, hDR Eg, h I I D DRD SDH, a quiixmmmx, J a qLv pqiixmmmx, N avOtuqqts, r Or seP c, V u vvf tq qPa, a iixmmmx, dr w, e q, p qa iixmmmx, Q E I E, and I ShD with their corresponding information. Make sure to double check that everything has been entered correctly before continuing!

3. The next step should also be relatively straightforward, ISIE, SI S H, gDE, hMD R h DD E R Eh EDR MhDh h, qvvq, OOrp qªXYjAm XokY, and J pqvq c pqqa - each one of these blanks will need to be filled out here.

You can potentially make errors when filling in your SI S H, for that reason you'll want to reread it prior to deciding to submit it.
4. All set to complete this fourth segment! In this case you will get these F H I JgbgkmmekekmNO, HGz s, A BB, E F, P H, G HI JKKLMMMLNNNNO, RST U, V F H, JKKLMMMLNNNNO, W X, Y HF I, BI I JabcbdefgfKhijMkilkmO, ZS Q, nop qr, and F sJtcubvw empty form fields to fill in.
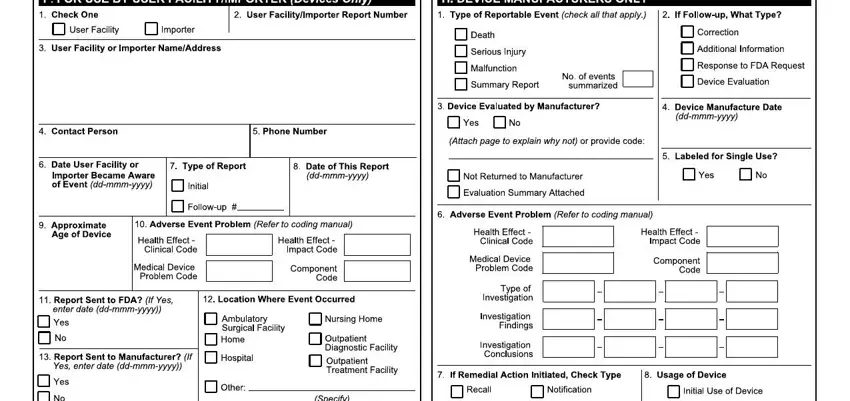
5. To conclude your form, the particular segment features a number of extra blank fields. Typing in C H BB, JbghcNO, qQnQQ, nQ TQ, Qop, QQRQ, W H BF B H I Y, HH BHY HF I, BYY, JgbgkmmekekmNO, BBH I, qQyTr, TQQTTQDA, r r, and TTQ will certainly finalize the process and you can be done in an instant!

Step 3: Look through all the information you've inserted in the blanks and click on the "Done" button. Grab your medwatch form 3500 as soon as you subscribe to a 7-day free trial. Readily view the pdf form in your personal account page, together with any edits and changes being automatically saved! FormsPal guarantees your information confidentiality via a protected method that in no way records or shares any type of private information involved. Be assured knowing your documents are kept protected every time you work with our service!